|
CHAPTER 2 GMO detection methods and validation
2.2 DNA-based methods
A review, with notes on future needs and directions.
Version June 22nd, 2001.
By
Arne Holst-Jensen, National Veterinary Institute, Section of Food & Feed Microbiology,
e-mail:
In this review the current methodological status of detection of GMO-derivatives in the food chain is presented. In reflection of the current state of the art the major focus will be on DNA-based methods, in particular those involving PCR (polymerase chain reaction). An introduction to the DNA molecule is linked as an annex, to help those readers unfamiliar with molecular biology to understand the technology of detection. The subheadings reflect the topics dealt with:
2.2.1 Genetically modified organisms (GMO)
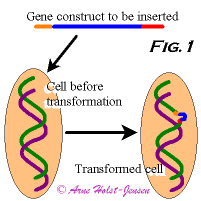
A genetically modified organism (GMO) is a living organism, e.g. a plant, whose genetic composition has been altered by means of gene technology. The genetic modification usually involves insertion of a piece of DNA (the insert), a synthetic combination of several smaller pieces of DNA, into the genome of the organism to be modified. This process is called transformation (Fig. 1). These smaller pieces of DNA are usually taken from other naturally occurring organisms. In bacteria and probably in plants in the near future, genetic modifications can also be introduced by altering existing codes without insertion of foreign DNA.
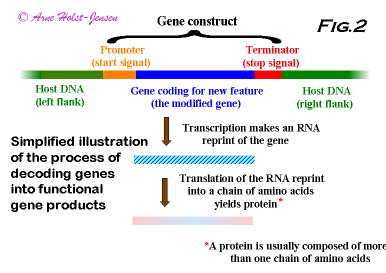
A typical insert (gene construct) in a GMO is composed of three elements: 1) The promoter element functions as an on/off switch for reading of the inserted/altered gene; 2) The gene that has been inserted/altered is coding for a specific selected feature; 3) The terminator element functions as a stop signal for reading of the inserted/altered gene. In addition several other elements can be present in a gene construct, and their function is usually to control and stabilize the function of the gene, demonstrate the presence of the construct in the GMO, or facilitate combination of the various elements of the construct. A gene construct must be integrated in the genome of the organism to become stably inherited. Therefore, the organisms own genome is also an important element. Figure 2 shows schematically the integration site of a gene construct and the various molecules derived from reading and translating the DNA.
2.2.2 Methods for detection of a GMO or its molecular derivatives
Detection of a GMO or a derivative of a GMO can be done by detecting a molecule (DNA, RNA or protein) that is specifically associated with or derived from the genetic modification of interest. The majority of the methods hitherto developed for detection of GMO and GMO-derivatives focus on detecting DNA, while only a few methods have been developed for detecting proteins or RNA. This has various reasons. DNA can be purified and multiplied in billions of copies in just a few hours with a technique called PCR (polymerase chain reaction, Fig. 3). Multiplication of RNA and proteins is a more complicated and slow process. DNA is a very stable molecule, while RNA is unstable. The stability of a protein varies and depends on the type of protein. There is normally a linear correlation between the quantity of GMO and DNA if the genetically modified DNA is nuclear, but not if it is extranuclear (in eukaryotes, chromosomal/extrachromosomal in prokaryotes). However, there is usually no such correlation between the quantity of GMO and protein/RNA. Finally, the genetic modification itself is done at the DNA level. At present, the genetically modified DNA is nuclear in all commercialised GMO.
Protein based methods rely on a specific binding between the protein and an antibody, a molecule related to those protecting our own organism against infections from bacteria and virus. The antibody recognizes the foreign molecule, binds to it, and in GMO detection assays the bound complex is successively detected in a chromogenic (colour) reaction. This technique is called ELISA (enzyme linked immunosorbent assays) and will not be described here. The antibody needed to detect the protein can not be developed without access to the purified protein. This protein can be purified from the GMO itself, or it can be synthesized in a laboratory if the composition of the protein is known in detail.
RNA based methods rely on specific binding between the RNA molecule and a synthetic RNA or DNA molecule (called a primer). The primer must be complementary to the nucleotide sequence at the start of the RNA molecule. The result is a double stranded molecule similar to DNA. Usually binding between the RNA molecule and the primer is followed by conversion of the RNA to a DNA molecule through a process called reverse transcription. Finally the DNA can be multiplied with PCR or it can be translated into as many as 100 copies of the original RNA molecule and the procedure can be repeated by using each copy as a template in a technique called NASBA (nucleic acid sequence-based amplification) that will not be described here. The specific primers needed for the procedure can not be developed without prior knowledge of the composition of the RNA molecule to be detected.
DNA based methods are primarily based on multiplying a specific DNA with the PCR technique (Fig. 3). Two short pieces of synthetic DNA (primers) are needed, each complementary to one end of the DNA to be multiplied. The first primer matches the start and the coding strand of the DNA to be multiplied, while the second primer matches the end and the non-coding (complementary) strand of the DNA to be multiplied. In a PCR the first step in a cycle involves separation of the two strands of the original DNA molecule. The second step involves binding of the two primers to their complementary strands, respectively. The third step involves making two perfect copies of the original double stranded DNA molecule by adding the right nucleotides to the end of each primer, using the complementary strands as templates. Once the cycle is completed, it can be repeated, and for each cycle the number of copies is doubled, resulting in an exponential amplification. After 20 cycles the copy number is approximately 1 million times higher than at the beginning of the first cycle. However, after a number of cycles the amount of amplification product will begin to inhibit further amplification, as will the reduction in available nucleotides and primers for incorporation into new amplification products. This effect is often referred to as the plateau effect.
One of the most commonly applied techniques for demonstration of the presence of a DNA or RNA is gel electrophoresis, a technique that allows the amount and size of the DNA/RNA to be estimated. This may eventually be coupled to digestion of the DNA with restriction enzymes that e.g. are known to cut a PCR fragment into segments of specified sizes. A more sophisticated technique involves determination of melting point profiles, by means of e.g. SYBR Green I, a dye that when intercalating double stranded DNA emits fluorescent light. When the temperature is increased, the DNA strands begin to separate. This leads to a corresponding reduction in fluorescence that can be measured directly e.g. on a real-time thermal cycler (PCR machine). The melting point is more characteristic of a specific DNA sequence than its size, but complete sequencing (determination of the order of nucleotides) of the DNA/RNA allows for more specific determination of the origin of the molecule. A fourth alternative is to use short synthetic molecules (similar to primers but called probes) and allow these to bind (hybridize) to the DNA/RNA. If appropriately designed, a probe will be able to discriminate between the correct molecule (sequence) and almost any other DNA/RNA molecule. Labelling of molecules with fluorescence, radioactivity, antibodies or dyes facilitate detection of the present molecules. For GMO analyses, gel electrophoresis and hybridisation techniques are currently the most commonly applied techniques.
2.2.3 The process of detection of a GMO or its molecular derivatives
The process of analysing for presence of GMO or GMO-derivatives in foodstuffs is stepwise (Fig. 4).
The first step is the sampling of the material to be analysed. Sampling strategies involves some rather complex statistics. Imagine a huge shipload of soybeans. How many beans do you need to sample and where do you take the samples from the load? Compare with canned foodstuffs in a grocery store. Do you need to take out every small piece of "something red" to analyse if it is derived from genetically modified tomato? If so, how do you do it? How many cans do you need to sample and analyse to produce a reliable estimate of the quantity that is genetically modified if you only find 3 tomatoes in each can in a batch of 25000 cans?
The second and third steps are the homogenizing of the sample and the isolation/purification of DNA/RNA/protein, respectively. The quality, purity and amount/concentration of the DNA/RNA/protein are the most critical factors determining the detection and quantification limits of the analyses. Again statistics are involved. How homogeneous must your sample be? What is the optimal particle size? How much material do you need? Do you need two, three, or fifty parallel isolations?
The fourth step is the analysis used to determine whether the molecule derived from the GMO is present. A range of alternative methods are available, with differences in their ability to discriminate between derivatives of different GMOs, and their reliability with respect to avoid false positive and negative results. The latter is very important. A method may not be capable of detecting the presence of GMO-derivatives although such are present in the sample (false negatives). Similarly, a method may report the presence of GMO-derivatives although such are NOT present in the sample (false poitives). How this is possible will be explained later. Other problems related to the method of analysis are: 1) Potentially the molecule to be detected has been (almost) completely degraded and is therefore no longer detectable, 2) Potentially refining steps in the processing of a foodstuff has removed (all of) the molecules which the detection method is designed to detect, 3) Potentially the analysis shall be performed on a mixture containing derivatives of several GMO and each must be identified.
The fifth step is the identification of the molecule that is detected. Whatever is detected, it may be a false positive. Consequently, it is usually necessary to verify the identity of the molecule.
The sixth step is optional quantification to determine how much genetically modified material the sample contains. Again, statistics are important and difficult. Quantification must always refer to something, e.g. "how much of the soya is genetically modified?" It is also difficult to determine the reliability of the method, and the uncertainty in the final estimates must be determined. How well a method performs must be tested through comparison with samples of known content, and preferably by independent comparison between several laboratories.
The seventh and final step is the interpretation of the analysis results, expressing any uncertainties associated with the result such as statistical inaccuracies, limitations of the method etc.
2.2.4 The current possibilities and limitations of the detection methods
The limits of detection and quantification are method specific but also depend on the sample that is being analysed. We can distinguish between three types of detection and quantification limits: 1) the absolute limits, i.e. the lowest number of copies that must be present at the beginning of the first cycle to obtain a probability of at least 95% of detecting/quantifying correctly, 2) the relative limits, i.e. the lowest relative percentage of GM materials that can be detected/quantified under optimal conditions, and 3) the practical limits, i.e. the limits applicable to the sample that is being analysed (taking into consideration the actual contents of the DNA sample and the absolute limits of the method) (Berdal & Holst-Jensen, in press).
With reference to figure 2, the specificity of currently available DNA based methods can be divided into four categories (Fig. 5): 1) screening methods that can detect a wide range of GMO without identifying the GMO, 2) screening methods that can detect a certain type of genetic modifications, 3) construct specific methods that sometimes can be used to identify the GMO, 4) transformation event specific methods that can be used to identify the GMO with special exceptions mentioned below. In addition PCR-based GMO analyses usually include testing for presence of DNA from the particular species of interest, e.g. soybean DNA. Sometimes (in the absence of amplifiable DNA from the particular plant species) GMO analyses also include testing for presence of amplifiable (multicopy) DNA from plants or Eukaryotes, e.g. chloroplast DNA or nuclear ribosomal small subunit genes (18S like).
The promoter and terminator elements used to transform most of the currently approved genetically modified plants are the Cauliflower Mosaic Virus promoter (P-35S) and the Agrobacterium tumefaciens nopaline synthase terminator (T-Nos). Although, other promoters and terminators have also been used, almost all GM plants contain at least one copy of the P-35S, T-35S and/or the T-Nos as a part of the gene construct integrated in its genome. Consequently, methods detecting one of these elements are popular for screening purposes (category 1 above). One problem with these methods is that the elements they detect are from naturally occurring virus and bacteria which are often present in fresh vegetables or the environment in which they are grown. Such elements therefore pose a significant risk of yielding false positive results.
The various genes inserted in a GMO may characterize a group of GMO, although they may not identify the GMO. Detection of the synthetic specific gene coding for the Bacillus thuringiensis endotoksin CryIA(b) demonstrates the presence of a genetically modified maize, but the gene has been used in more than one GMO. Consequently, category 2 methods like those that can detect the CryIA(b) gene can tell us more than the category 1 screening methods, but will not be suitable for identification of the specific GMO.
The synthetic CryIA(b) gene has been integrated with different specific regulatory elements (promoters and terminators) in the various GMO containing the gene. Currently it is therefore possible to identify the GMO with category 3 detection methods targeting the junctions where the gene and regulatory elements are fused. However, in the future, even these junctions may be found in more than one GMO.
With currently available technology it has not yet been possible to control where in the genome of a plant the insert is integrated. If the same insert is integrated into the genome of the same type of organism several times, the likelihood of integration of the insert in the same place in the genome two times is usually negligible. Consequently, the junction between the integration site and the insert will be unique for each transformation event. Category 4 methods detect these regions and will remain specific for the transformation even when the same construct has been integrated into the same plant species many times. However, category 4 methods are under development, and none has yet been published.
When two GMO are crossed, e.g. two different approved genetically modified maize cultivars, the resulting hybrid offspring may possess the genetic modifications from both parent cultivars. This phenomenon is called "gene stacking". In the U.S.A. this type of hybrid GMO is not regulated, because both parent cultivars are approved. In the European Union, however, the hybrid is considered to be a new GMO and requires separate approval. None of the above four categories of analysis methods will be able to identify cases of gene stacking. Instead, cases of gene stacking will give results indistinguishable from the separate detection and identification of each of the parental cultivars in the sample.
Table 1. Examples of published methods to detect GMO derivatives grouped according to categories of specificity (see text for details).
| Type of method |
Target sequence |
Reference |
|
|
|
| Method to detect plant derived DNA |
|
|
|
Chloroplast tRNALeu gene (trnL) intron |
Taberlet & al., 1991 |
| Methods to detect specific plant species |
|
|
|
Corn/maize single copy invertase gene |
Ehlers & al., 1997 |
|
Soybean single copy lectin gene |
Meyer & al., 1996 |
|
Tomato single copy polygalacturonase gene |
Busch & al., 1999 |
| Screening methods (category 1) |
|
|
|
Cauliflower mosaic virus promoter (P-35S) |
Pietsch & al., 1997 |
|
Nopaline synthase terminator (T-Nos) |
Pietsch & al., 1997 |
| Gene specific methods (category 2) |
|
|
|
bar (phosphinotricin acetyltransferase) gene |
Ehlers & al., 1997 |
|
CryIA(b) gene (synthetic) |
Ehlers & al., 1997 |
|
|
Vaïtilingom & al., 1999 |
| Construct specific methods (category 3) |
|
|
|
Bt11 maize: junction alcohol dehydrogenase 1S intron IVS6 (enhancer) - CryIA(b) gene |
Matsuoka & al., 2001 |
|
Bt176 maize: junction CDPK (calcium dependent protein-kinase) promoter - synthetic CryIA(b) gene |
Hupfer & al., 1998 |
|
GA21 maize: OTP (enhancer) - epsps gene (RoundupReady tolerance) |
Matsuoka & al., 2001 |
|
Mon810 maize: junction P-35S - heat shock protein (hsp) 70 intron I (enhancer) |
Zimmermann & al., 1998 |
|
Mon810 maize: junction hsp 70 intron - CryIA(b) gene |
Matsuoka & al., 2001 |
|
RoundupReady®: junction P-35S - Petunia hybrida CTP (chloroplast transit peptide) |
Wurz & Willmund, 1997 |
|
T25 maize: junction pat (phospinotricin acetyltransferase) gene - T-35S |
Matsuoka & al., 2001 |
|
Zeneca tomato: junction T-Nos - truncated tomato polygalacturonase gene |
Busch & al., 1999 |
| Event specific methods (category 4) |
|
|
|
Bt11 maize: junction host plant genome - integrated recombinant DNA |
Zimmermann & al., 2000 |
|
RoundupReady® soybean: junction host plant genome - integrated recombinant DNA |
Berdal & Holst-Jensen, in press |
|
|
Taverniers & al., in press |
|
|
Terry & Harris, in press |
2.2.5 Multiplex PCR-based detection methods
With multiplex PCR-based methods several target DNA sequences can be screened for and detected in a single reaction. Although in principle, standard PCR methods may be combined in the same reaction, in practice this will often create an unacceptably high risk of producing incorrect results from analyses of real samples. Firstly, each method may require different reaction conditions, e.g. different temperature regimes or different reagent concentrations. Secondly, the combination of primers from different methods may increase the risk of amplifying DNA fragments other than the target fragments. Thirdly, when more than one target fragment is being amplified in a PCR reaction, the two fragments (amplicons) will compete for reagents etc. Normally, a fragment present in a large number of starting copies will outcompete another fragment that may be present only in very few numbers. However, if two amplicons are amplified with significantly different efficiency this may also have a severe impact on the final ratio of the two amplicons, e.g. if the starting copy numbers were more or less the same for both. Consequently, development of multiplex assays requires careful testing and validation. After the PCR the resulting pool of amplified DNA fragments needs to be further analysed to distinguish between the various amplicons. This may be done by the use of specific hybridisation probes (possibly also during PCR in real-time assays), by gel electrophoresis and comparison of fragment sizes or by the use of specifically labelled primers. While several research groups are currently developing a number of multiplex assays, hitherto only one paper has been published presenting a multiplex assay for detection of five GM-maize (Bt11, Bt176, Mon810, T25 and GA21; Matsuoka & al, 2001).
The advantage of multiplex methods is evidently that fewer reactions are needed to test a sample for potential presence of GMO-derived DNA. In particular, if further quantitation is needed, it may be of importance to know which GMO to quantify since a quantitative PCR is relatively expensive. Identification of each specific GMO may also be helpful to identify whether detected DNA is derived from approved or non-approved GMO.
2.2.6 Quantitation methods using PCR
In principle, PCR based quantitation can be performed either after completion of the PCR (end-point analysis), or during the PCR (real-time analysis).
End-point analyses are usually based on comparison of the final amount of amplified DNA of two DNA targets, the one to be quantified and a competitor (an artificially constructed DNA that is added in a small and known quantity prior to the PCR amplification and that is coamplified with the target that is to be quantified). This is called competitive quantitative PCR (see Fig. 6), and it requires that the two DNA targets are amplified with equal efficiency since otherwise the final amount of product is not linearly correlated with the starting amount.
A dilution series of the DNA to be analysed is prepared, and a constant amount of the competitor is added. After completion of the PCR the resulting amplification products are visualized through gel electrophoresis and when both DNA targets yield the same amount of product it is assumed that the starting amount was also the same. By setting up two competetive PCRs, one for the GMO (e.g. RoundupReady soybean) and one for the species of interest (e.g. soybean), and including competitors in both, the quantity of GMO relative to the species can be estimated by extrapolation from the degree of dilution and concentration of the competitors. Such assays are referred to as quantitation by double competitive PCR.
In real-time analyses the amount of product synthesised during PCR is estimated directly by measurement of fluorescence in the PCR reaction. Several types of hybridisation probes are available that will emit fluorescent light corresponding to the amount of synthesized DNA. However, the amount of synthesised product can also be estimated with fluorescent dyes, e.g. SYBR Green I that intercalates double-stranded DNA. With the latter, it is not possible to distinguish between the specific product and non-specific products, and consequently the use of specific hybridisation probes is normally preferred. As with double competitive PCR, the quantitative estimate is based on extrapolation by comparison of the GMO sequence relative to the reference of interest (Fig. 7). The idea is that with the use of fluorescence it becomes possible to measure exactly the number cycles that are needed to produce a certain amount of PCR product. This amount corresponds to the amount producing a fluorescence signal clearly distinguishable from the background signal and measured well before the plateau effect becomes a problem. The number is called the Ct-value. Then by comparison of Ct-values for the GMO target sequence, e.g. RoundupReady soybean 3' integration junction, and the reference gene, e.g. soybean lectin, it becomes possible to estimate the ratio of the GMO target sequence to the reference sequence in terms of difference in number of cycles needed to produce the same quantity of product. Since one cycle corresponds to a doubling of the amount of product, a simple formula can be presented to estimate the ratio in percent (see Fig. 7). While real-time PCR requires more sophisticated and expensive equipment than competitive PCR, it is faster and (at least sometimes) more specific. Although what is said above may give the impression that quantitation can be done directly from comparison of Ct-values, most laboratories now adjust their estimates by comparison to a standardcurve, i.e. a series of measurements from a DNA series with known GMO content.
Table 2. Examples of published methods to quantify GMO derivatives grouped into competitive and real-time PCR methods.
| Type of method |
Target GMO |
Reference |
| Double competitive PCR methods |
|
|
|
RoundupReady soybean (raw materials) |
Van den Eede & al., 2000 |
|
Bt176 maize (raw materials) |
Van den Eede & al., 2000 |
|
P-35S (screening) |
Hardegger & al., 1999 |
|
T-Nos (screening) |
Hardegger & al., 1999 |
|
Epsps gene (RoundupReady) |
Studer & al., 1998 |
|
Synthetic CryIA(b) gene |
Studer & al., 1998 |
|
Bt-11 maize (event specific) |
Zimmermann & al., 2000 |
| Real-time PCR methods |
|
|
|
RoundupReady soybean (event specific) |
Berdal & Holst-Jensen, in press |
|
|
Taverniers & al, in press |
|
|
Terry & Harris, in press |
|
Synthetic CryIA(b) gene |
Vaïtilingom & al, 1999 |
2.2.7 Future needs and directions
The current European regulations require labelling of a foodstuff as being "from genetically modified...." if more than 1% of an ingredient is derived from GMO and this can be detected in the foodstuff. However, the analysis methods can not distinguish between two different ingredients derived from the same species in a single foodstuff, e.g. soy protein and soy flour in one product. The methods can only be used to detect and quantify the content of GMO at the species level. For quantitation this means that the quantity must be expressed as the amount of genetically modified soy out of total soy in the foodstuff, or at the product level as the amount of genetically modified soy in the foodstuff (without reference to the total amount of soy in the foodstuff).
If a food product contains "gene stacked" GMO, then quantification may become confusing. If for example, 0.75 % of the maize is from a GMO derived from a cross between two different maize transformants, quantification will report 1.5 % GMO maize. The current European regulations do not specify how this type of case shall be handled.
Until now it has been possible for the companies seeking approval of their GMO in Europe to request that sequence information in the dossiers describing the GMO be kept confidential. Consequently, unavailability of sequence data has made it difficult for many laboratories to develop detection methods. Similarly, it has been difficult to obtain material of the majority of GMO (approved as well as non-approved). Such material is absolutely necessary to validate the detection methods. A collaborative attitude from the GMO companies to competent authorities and control laboratories, the recent revision of EC directive 90/220 (new EC directive 18/2001) and ongoing revisions of other EC directives and regulations, may lead to better access to sequence information and GMO material in the future.
Better methods for isolation of the molecules to be detected, and for qualitative and quantitative analysis of GMO are needed and under development. The improvements will include better statistics, better sampling, adaptations to specific types of foodstuffs, higher specificity with respect to identification of the GMO, faster analyses, reduced costs, reductions in inaccuracies, etc. Several projects, e.g. the EC supported shared cost research and technological development project "Qpcrgmofood" (Reliable, standardised, specific, quantitative detection of genetically modified food; contract no. QLK1-1999-01301; http://www.vetinst.no/) are working on this. The majority of developments are on DNA methods.
Increased specificity may be obtained with 1) PCR methods targeting the junction between integration site and insert DNA, 2) GMO specific fingerprinting methods similar to those used to identify criminals, 3) diagnostic microchips similar to those used to determine if a person is predisposed for inheritable diseases. Other alternatives are currently being examined.
Today, each GMO must be identified separately. If all GMO present in a sample can be detected in a single reaction, analyses will be faster and costs potentially reduced.
Calibration of quantitative assays suffers from the lack of suitable calibrants. Although certified reference materials are available for some GMO, these reference materials should preferably not be used to calibrate the quantitative assays because it may be possible to alter the relative ratio of the GMO to non-GMO during the processing of the material (see chapter 2.4). A calibrant is a pure DNA sample with a specific ratio of GMO to non-GMO. Calibrants will hopefully be available in the near future.
Only a few of the presently available methods have been validated in collaborative trials (ring-trials with several independent laboratories). Validation with different types of foodstuffs also remains to be assessed for the majority of methods. Such validation is needed before the methods can be considered truly reliable.
Finally, GMO analysis laboratories should participate in proficiency tests organised by independent bodies, to regularly test and demonstrate that their analyses are reliable and accurate. Since late in 1999 proficiency tests have been organised by U.K. authorities: the Food Standards Agency, and the Central Science Laboratory (FAPAS). For industry, control authorities and others purchasing GMO analyses, it is highly recommendable to require from the laboratories that they are accredited for e.g. PCR based diagnostics, that they participate in proficiency tests, and that the laboratories also make public how they perform in these tests. Preferably, and if possible, the analysis methods should be international standards (not yet available but in preparation), to avoid disputes between parties using different methods.
References:
Berdal, K.G. & A. Holst-Jensen (in press). RoundupReady® soybean event specific real-time quantitative PCR assay and estimation of the practical detection and quantification limits in GMO analyses. Eur. Food Res. Technol.
Busch, U., B. Mühlbauer, M. Schulze & J. Zagon (1999). Screening- und spezifische Nachweismethode für transgene Tomaten (Zeneca) mit der Polymeraskettenreaktion. Deutsche Lebensmittelrundschau, Heft 2: 52-56.
Ehlers, B., E. Strauch, M. Goltz, D. Kubsch, H. Wagner, H. Maidhof, J. Bendiek, B. Appel & H.-J. Buhk (1997). Nachweis gentechnischer Veränderungen in Mais mittels PCR. Bundesgesundhbl. 4: 118-121.
Hardegger, M., P. Brodmann & A. Herrmann (1999). Quantitative detection of the 35S promoter and the NOS terminator using quantitative competitive PCR. Eur. Food Res. Technol. 209: 83-87.
Hupfer, C., H. Hotzel, K. Sachse & K.-H. Engel (1998). Detection of the genetic modification in heat treated products of Bt maize by polymerase chain reaction. Lebensm. Unters. Forsch. 206(A): 203-207.
Matsuoka, T., H. Kuribara, H. Akiyama, H. Miura, Y. Goda, Y. Kusakabe, K. Isshiki, M. Toyoda & A. Hino (2001). A multiplex PCR method of detecting recombinant DNAs from five lines of genetically modified maize. J. Food Hyg. Soc. Japan 42: 24-32.
Meyer, R., F. Chardonnens, P. Hübner & J. Lüthy (1996). Polymerase chain reaction (PCR) in the quality and safety assurance of food: detection of soya in processed meat products. Z. Lebensm. Unters. Forsch. 203: 339-344.
Pietsch, K., H.U. Waiblinger, P. Brodmann & A. Wurz (1997). Screeningverfahren zur Identifizierung "gentechnisch veränderter" pflanzlicher Lebensmittel. Deutsche Lebensm. Rundsch. 93: 35-38.
Studer, E., C. Rhyner, J. Lüthy & P. Hübner (1998). Quantitative competitive PCR for the detection of genetically modified soybean and maize. Z. Lebensm. Unters. Forsch. A. 207: 207-213.
Taberlet, P., L. Gielly, G. Pautou & J. Bouvet (1991). Universal primers for amplfication of three non-coding regions of chloroplast DNA. Plant Mol. Biol. 17: 1105-1109.
Taverniers, I., P. Wiendels, E.Van Bockstaele & M. De Loose (in press). Use of cloned DNA fragments for event specific quantification of genetically modified organisms in pure and mixed food products. Eur. Food Res. Technol.
Terry, C. & N. Harris (in press). Event specific detection of RoundupReady soya using two different real time PCR detection systems. Eur. Food Res. Technol.
Vaïtilingom, M., H. Pijnenburg, F. Gendre & P. Brignon. Real-time quantitative PCR detection of genetically modified maximizer maize and RoundupReady soybean in some representative foods. J. Agric. Food Chem. 47: 5261-5266.
Van den Eede, G., M. Lipp, F. Eyquem & E. Anklam (2000). Validation of a double competitive polymerase chain reaction method for the quantification of GMOs in raw materials. Report published by the European Commission, Joint Research Centre, IHCP, Ispra, Italy. EUR 19676, 40pp.
Zimmermann, A., M. Liniger, J. Lüthy & U. Pauli (1998). A sensitive detection method for genetically modified MaisGard" corn using a nested PCR-system. Lebensm.-Wiss. u. Technol. 31: 664-667.
Zimmermann, A., J. Lüthy & U. Pauli (2000). Event specific transgene detection in Bt11 corn by quantitative PCR at the integration site. Lebensm.-Wiss. u. Technol. 33: 210-216.
|
{kind=link}
{kind=link}